 腾讯登录
腾讯登录美国西北大学与大连医科大学:重大进展!发现肿瘤发生新途径,揭示癌症治疗新靶点
| 导读 | 我们发现p23是COX-2先前未被发现的转录因子,其核定位预示着不良的临床结果。 |
肿瘤内琥珀酸盐促进p23在K7、K33和K79位点的琥珀酸化,从而驱动其核易位进行COX-2转录,从而促进肿瘤生长。然后,我们通过虚拟和生物联合筛选,从160万种化合物中鉴定出M16是一种有效的p23琥珀酰化抑制剂。M16抑制p23琥珀酰化和核易位,以p23依赖性方式减弱COX-2转录,显著抑制肿瘤生长。因此,我们的研究将p23定义为肿瘤进展中琥珀酸激活的转录因子,并为抑制p23琥珀酸化作为抗癌化疗提供了理论依据。

https://www.science.org/doi/10.1126/sciadv.ade0387
研究背景
01
慢性炎症促使大多数癌症的发生和恶性发展。肿瘤相关炎症分为外源性炎症和内源性炎症。肿瘤外源性炎症是由多种炎症因子引起的,如肿瘤坏死因子-α (TNF-α)、白细胞介素6 (IL-6)和IL-1β,这些炎症因子是由巨噬细胞等炎症细胞分泌的。相比之下,内在炎症可由促进炎症细胞募集和激活的癌症遗传和代谢改变触发。外在和内在炎症途径都创造了一个有利于癌症生长和重新编程癌细胞代谢的微环境。另一方面,癌细胞代谢的改变导致内源性代谢物(如前列腺素和琥珀酸盐)的产生加剧,同时引发肿瘤微环境中的外源性和内源性炎症。例如,环氧化酶2 (COX-2),是一种催化前列腺素产生的酶,可以复原免疫抑制细胞,包括调节性T细胞和B细胞。进入肿瘤组织,诱导肿瘤环境中的免疫抑制状态,有利于癌细胞的存活和增殖。此外,COX-2还能诱导癌症干细胞样活性,促进癌细胞的凋亡抵抗、血管生成、侵袭和转移。肿瘤微环境中前列腺素和琥珀酸盐的异常积累以及COX-2表达的升高通常与许多类型癌症的不良临床结果相关。COX-2表达可被炎性细胞因子,包括TNF-α、IL-1β和IL-6,以及其他刺激,如脂多糖(LPS),通过核因子κB (NF-κB)信号传导迅速诱导。针对外在和内在炎症因子抑制COX-2表达的治疗策略已被证明是一种很有前途的癌症治疗方法。然而,肿瘤代谢物是否以及如何直接参与肿瘤发生和发展过程中促进COX-2的表达尚不清楚。
由PTGES3(前列腺素E合成酶3)基因编码的分子伴侣蛋白p23是一种高度保守的蛋白,在从酵母到人类的真核生物中表达。p23被认为是热休克蛋白90 (Hsp90)的共同伴侣,在稳定Hsp90复合物与几种客户,包括雌激素受体,雄激素受体和端粒酶。值得注意的是,p23上调已在多种类型的癌症中被观察到,包括前列腺癌和乳腺癌,以及急性淋巴细胞白血病。p23的过表达(OE)增强细胞迁移,增加雄激素受体蛋白水平和活性,促进癌症发展,并与乳腺癌患者预后不良相关。此外,它还与肿瘤细胞耐药增加有关。越来越多的证据表明,p23也以不依赖于hsp90的方式发挥其部分功能。然而,不依赖于hsp90的p23功能与癌症之间的关系以及p23如何实现其不依赖于hsp90的功能的分子机制仍然是一个生物学之谜。
出乎意料的是,我们发现p23作为一组肿瘤驱动基因表达的转录因子,包括COX-2和趋化因子CXCL1、CXCL8和CCL20。p23转录激活是由肿瘤代谢物琥珀酸的异常积累引发的,通过促进其琥珀酰化来促进核易位。药理抑制p23琥珀酰化可显著降低p23靶基因表达,抑制肿瘤生长。因此,我们的研究结果重新定义了p23是一个不依赖于hsp90的,能促进肿瘤生长的COX-2转录因子。
研究成果
02
核p23通过调节COX-2转录促进肿瘤增殖
为了探索p23在肺腺癌中的重要致病作用,我们通过组织芯片检测了86例肺腺癌患者肿瘤和邻近组织中p23的表达。免疫组化(IHC)染色和Western blotting都检测到肿瘤中p23的表达比邻近的正常组织显著增加。值得注意的是,IHC染色还检测到人类原发性肺癌组织中明确的p23核定位,亚细胞分离和Western blotting分析进一步证实了这一点。定量分析显示,超过90%的肿瘤组织具有p23核表达,而邻近组织中只有约5%,这表明p23的表达和核积累在肺腺癌中升高。同样,与对照组正常人肺成纤维细胞(HLF)细胞相比,在多种人肺癌细胞系中也观察到p23上调及其核定位。临床病理特征分析显示,高核p23表达与肿瘤分期晚期和无病生存期降低相关。这些结果表明,p23表达的升高及其核定位预示着肺腺癌的临床预后较差,并可能与肿瘤发生有关,这与以往的研究一致,表明p23在肿瘤中增加并在核中积累。
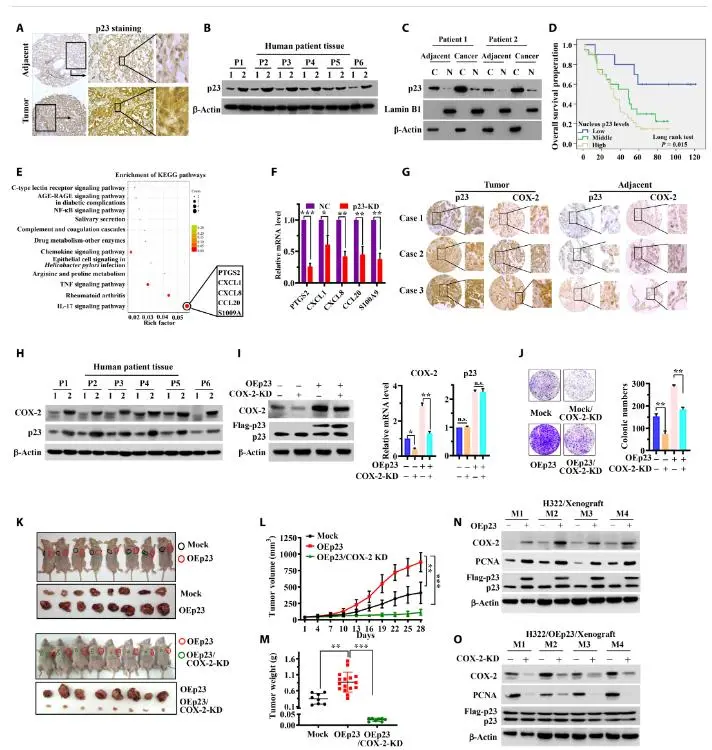
核p23通过调节COX-2转录促进癌症进展
进一步分析表明,通过短发夹介导的RNA (shRNA)介导的p23- kd敲低,内源性p23耗损显著降低了A549肺癌细胞的增殖、侵袭能力和集落形成。进一步分析表明,p23抑制导致细胞周期阻滞在S期,并使肺癌细胞对顺铂(CDDP)诱导的凋亡敏感。此外,Western blotting分析发现,参与细胞存活和细胞周期进程的蛋白表达减少,但细胞凋亡因子的表达增加。相比之下,p23稳定的OE在肺癌细胞中以相反的方式改变了参与细胞生长和死亡的蛋白表达。这些数据表明p23是促进肺癌细胞生长和抑制其凋亡的致癌因子。
为了确定参与p23驱动肺癌进展的分子途径,我们进行了RNA测序(RNA-seq)分析,以探索p23缺失后明显改变的基因。从所有三个实验组中鉴定出92个差异表达基因(DEGs), p23缺失增加了两倍以上或减少了50%。逆转录定量聚合酶链反应(RT-qPCR)分析证实了所有8个随机选择的deg,为我们的RNA-seq分析结果提供了高可信度。92个deg的KEGG分析表明,p23通过多种炎症相关途径促进肺癌的进展,如IL-17、TNF、趋化因子和NF-κB。特别是,炎症因子和趋化因子,包括S100A9、CCL20、PTGS2、CXCL1和CXCL8的表达因p23抑制而降低。在92个DEGs中,编码COX-2的PTGS2基因因p23抑制而减少最多。
COX-2是促进肿瘤生长的重要炎症驱动因子。我们随后提出p23至少部分通过调节COX-2表达参与肿瘤发生。通过免疫组化染色,p23和COX-2在肺肿瘤组织中的表达均明显高于邻近正常组织。COX-2与p23核呈显著正相关。Western blotting进一步证实了p23和COX-2在原发性肺癌和癌旁组织以及多种癌细胞系中的表达趋势相似。此外,在不同类型的肿瘤细胞中,p23缺失显著下调COX-2的表达,表明p23在肺癌细胞中调节COX-2的表达。
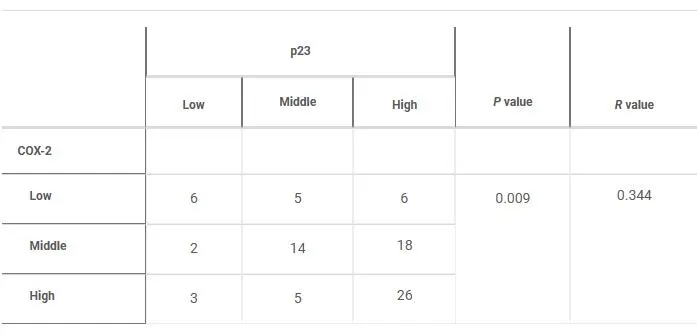
肺腺癌标本中p23核与COX-2蛋白表达呈正相关
核p23作为COX-2的转录因子,促进肿瘤生长
我们进行了一次/氢氘交换(HDX) - ms分析使用串行合成肽覆盖96%的p23蛋白质识别蛋白质区域负责p23-DNA绑定。HDX的p23绑定cox - 2启动子DNA片段(CPDF)明显降低肽90年到99年,而其他的肽没有,甚至肽42至52 HDX率增加,表明残留90 - 99负责p23 CPDF绑定。我们进一步进行分子动力学(MD)模拟来探索p23与CPDF结合的动力学。均方根偏差(RMSD)值表明p23/CPDF在100 ns后达到平衡, p23与CPDF保持稳定的结构,并且在MD模拟中它们的结合亲和力更强。p23的正电荷残基R88、R93和K95很大程度上归因于与COX-2启动子DNA的重要结合。这三个氨基酸残基中的每一个突变为丙氨酸都足以减少p23与COX-2启动子的结合,从而消除p23诱导的COX-2报告子活性。此外,如前所述,我们还使用锚离系统来探索p23调制COX-2的需求。结果表明,Flag-p23-FRB能有效促进COX-2的表达,而在雷帕霉素存在下,COX-2的表达明显降低。这些数据进一步证实了我们p23核易位促进COX-2基因转录的初步结论。综上所述,这些结果表明p23是先前未被发现的肺癌细胞中COX-2表达的转录因子。
琥珀酰化对p23的转录活性至关重要
蛋白质琥珀酰化通常在高琥珀酰辅酶A (CoA)浓度下通过非酶机制发生。琥珀酰辅酶a体外培养诱导H322/p23- oe细胞p23超琥珀酰化,体外纯化重组p23。为了鉴定p23琥珀酰化的残基,将rp23与琥珀酰辅酶a孵育,然后进行质谱分析。通过蛋白质组学分析,我们在p23中发现了三个潜在的琥珀化赖氨酸位点(K7、K33和K79),这些位点在小鼠和人类中高度保守。通过Arg置换的P23突变体(KR突变)分别在这三个位点上或两个位点上或全部位点上产生。出乎意料的是,p23的K79突变体,包括K79R, K7R/K79R, K33R/K79R,或K7R/K33R/K79R,与p23-wt(野生型)或其他突变体相比,蛋白质水平降低,这表明K79对于维持p23蛋白的稳定性至关重要,因为琥珀酰化已被证明可以减少溶酶体蛋白的降解。然而,K7或K33赖氨酸残基或两者的突变显著降低了p23琥珀酰化水平及其核易位,而没有改变p23蛋白的总表达,因此,COX-2报告基因检测和COX-2蛋白表达表明,p23转录活性受损。琥珀酰辅酶a体外培养明显促进了重组wt p23蛋白的琥珀酰化,但对其K7R/K33R突变体没有作用。总之,这些结果表明K7/K33琥珀酰化驱动p23核易位以表达COX-2基因。
总之,我们通过促进COX-2表达发现了一个以前未知的肿瘤发生炎症途径,这为在抗肿瘤治疗中操纵p23转录激活提供了理论依据。通过使用计算化学方法建立针对“p23琥珀酰化-COX-2启动子结合”的虚拟药物筛选模型,我们对160万种化合物进行了系统的计算机筛选,并随后进行了验证分析,发现M16是一种有效的p23抑制剂,可以有效地减弱p23的核易位,同时阻断其与COX-2启动子的结合。与经典的COX-2活性抑制剂CB协同作用,M16治疗显著抑制小鼠体外肿瘤生长,无任何可检测到的急性毒性。值得注意的是,在A549细胞中,M16明显抑制p23琥珀酰化,但不影响p65磷酸化水平,而CB明显抑制p65磷酸化,但不影响p23琥珀酰化,并且两种药物联合抑制NF-κB途径和p23琥珀酰化。这些结果证实了我们最初的结论,即M16和CB协同增强抗肿瘤活性,至少部分是通过抑制COX-2的表达。因此,我们的研究结果揭示了分子伴侣p23作为一种肿瘤促进转录因子,并为炎症性疾病和癌症的治疗提供了一个以前未知的治疗靶点。分子伴侣p23除了在肿瘤发生中发挥作用外,还可能作为调节关键生物学功能的转录因子。未来的研究需要解剖p23转录因子的细胞、组织或器官特异性功能。(转化医学网360zhyx.com)
参考资料:
https://www.science.org/doi/10.1126/sciadv.ade0387
注:本文旨在介绍医学研究进展,不能作为治疗方案参考。如需获得健康指导,请至正规医院就诊。










还没有人评论,赶快抢个沙发