 腾讯登录
腾讯登录2023最新!北京大学汤富酬等团队发布单细胞测序技术最新研究成果
| 导读 | 基于第三代测序(TGS)平台的单细胞RNA-seq技术的进步加速了生物学研究。自2016年以来,已经开发了几种基于TGS平台的单细胞RNA-seq方法。受低准确度和灵敏度的限制,它们要么结合基于NGS平台的方法降低错误率,要么牺牲通量来提高检测率。 |
2023年1月11日,北京大学汤富酬等团队合作在《Cell Discovery》在线发表题为“High-throughput and high-sensitivity full-length single-cell RNA-seq analysis on third-generation sequencing platform”的研究论文,该研究在第三代测序平台上进行了高通量、高灵敏度的全长单细胞RNA-seq分析。

https://www.nature.com/articles/s41421-022-00500-4
研究概述
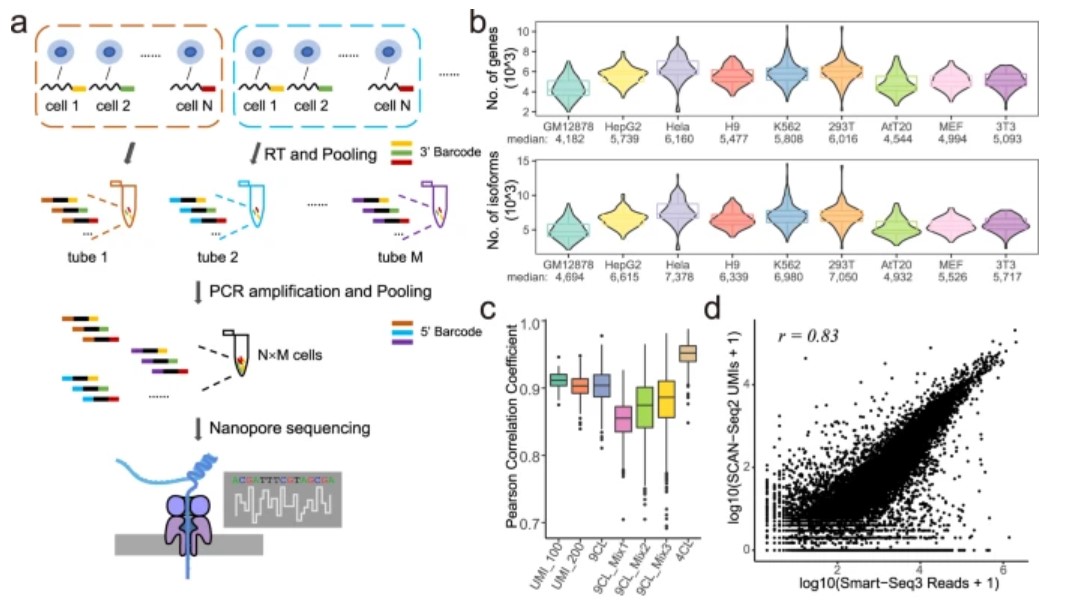
为了克服这些缺点,研究人员开发了SCAN-seq2,这是一种基于TGS平台的高通量,高灵敏度的全长RNA-seq方法。他们对来自九个细胞系的总共5472个细胞进行了SCAN-seq2,显示了数据处理管道的详细流程图以及过滤、解复用和重复数据删除后读取的总体统计数据。
通过参考引导的转录组组装,研究人员鉴定了数千种新型全长RNA亚型。假基因的转录本可以与相应亲本基因的转录本区分开来,其中数百个显示出细胞类型特异性表达模式。此外,研究发现可以准确确定高度多态性T细胞受体(TCR)和B细胞受体(BCR)基因(免疫球蛋白)的V(D)J重排事件。最后,研究证明了HepG2和Hela细胞在剪接体抑制剂异银杏素(IGG)处理后的保守凋亡反应。SCAN-seq2被证明是单细胞全长转录组研究的一种新的有前途的工具。
研究进展
在SCAN-seq2中,每32个具有不同3 '条形码的单细胞的第一链cDNAs在逆转录后聚集在一起。通过在PCR引物中引入24-nt条形码,在PCR扩增过程中加入不同的5 '条形码。通过这种方法,该研究一次测序最多可以测序3072 (32 × 96)个单细胞。平均而言,当对960个细胞进行测序分析时,研究人员在每个细胞中检测到超过4000个基因和4500个组装良好的RNA异构体。每个24 bp条形码的序列错误数总体上小于3,远低于96个不同条形码的编辑距离(11个碱基)。
研究还比较了通过SCAN-seq2和基于NGS的方法鉴定的同一文库的ERCC UMI。当编辑距离为1时,86%的SCAN-seq2 UMI可以分配给相应的NGS UMI,验证了SCAN-seq2 UMI的合理质量。这些结果表明,SCAN-seq2具有高通量和高灵敏度。
然后,研究人员评估了交叉污染的水平。经过严格的质量控制,来自Library 4CL的所有细胞的读数基本上与小鼠或人类参考转录组正确比对,只有一个细胞(0.28%)被鉴定为受污染,证实了SCAN-seq2的低交叉污染率。添加 UMI 后,SCAN-seq2 实现了与黄金标准方法 Smart-seq3 相当的精度。在所有5个文库中,UMI计数与实际ERCC浓度之间的Pearson相关系数均高于0.85。SCAN-seq2和Smart-seq3合并数据对全局基因表达的相关性为0.83,验证了这两种方法的一致性。此外,每对单个细胞之间的平均相关性达到0.95,与Smart-seq3一样高,表明SCAN-seq2具有很高的重现性。
癌细胞系中异银杏素(IGG)反应的SCAN-seq2技术性能和分析
SCAN-seq2也可用于研究TCR和BCR,其中转录本是在可变(V),多样性(D)和连接(J)基因片段重组后产生的。在H9和GM12878中均检测到大量抗原受体编码转录本。对于TCR,在H9细胞中仅检测到基因TRAC和TRBC1的转录本,表明TCRαβ基因的表达。我们发现基本上所有的H9细胞对β链都具有相同的V(D)J重排,TRBV13作为V元素,TRBD263作为D元素,TRBJ1-2作为J元素。正如预期的那样,没有检测到α链的D元素。进一步鉴定了两个亚克隆,对应于α链的两个VJ重排。GM12878的BCRs亚克隆之间的主要区别在于重链,而轻链基本相同。我们鉴定的最大GM12878克隆与GM12878免疫球蛋白的“真实基因注释”完全一致5.这些结果证明,SCAN-seq2可以准确确定恒定区域的TCR和BCR基因转录本和V(D)J重组,从而可以在单细胞分辨率下准确检测T细胞和B细胞的克隆多样性。
研究意义
总而言之,与其他已发表的基于TGS平台的scRNA-seq方法相比,SCAN-seq2同时表现出高通量和高灵敏度。作为一种全长测序方法,SCAN-seq2可以获得更均匀的转录本信息,而不会发生片段和富集。因此,为了获得全长转录本每个片段的相同覆盖率,SCAN-seq2所需的测序深度可能远低于短读长测序。作为一种更方便的工具,SCAN-seq2可用于以单细胞和单个RNA亚型分辨率研究不同的生物系统,并帮助了解许多疾病的复杂机制。(转化医学网360zhyx.com)
参考资料:
https://www.nature.com/articles/s41421-022-00500-4
注:本文旨在介绍医学研究进展,不能作为治疗方案参考。如需获得健康指导,请至正规医院就诊。










还没有人评论,赶快抢个沙发