 腾讯登录
腾讯登录长垣——华大基因新生儿遗传代谢病检测“筛诊一体”的先行者
| 导读 | 国家卫生健康委员会发布健康儿童行动计划 (2018-2020) |
引 言
“儿童健康是全民健康的基础,
是经济社会可持续发展的重要保障。”
国家卫生健康委员会发布健康儿童行动计划 (2018-2020)
新生儿遗传代谢病“筛诊一体”的先行者
华大基因新生儿遗传代谢病检测 (简称“新筛”) “筛诊一体”指将串联质谱与测序两种检测技术联合,帮助新筛疑似阳性患儿尽快对疾病进行确诊,真正做到对新生儿遗传代谢病的早发现、早诊断、早治疗! 避免或减轻儿童致残,减少社会经济负担。
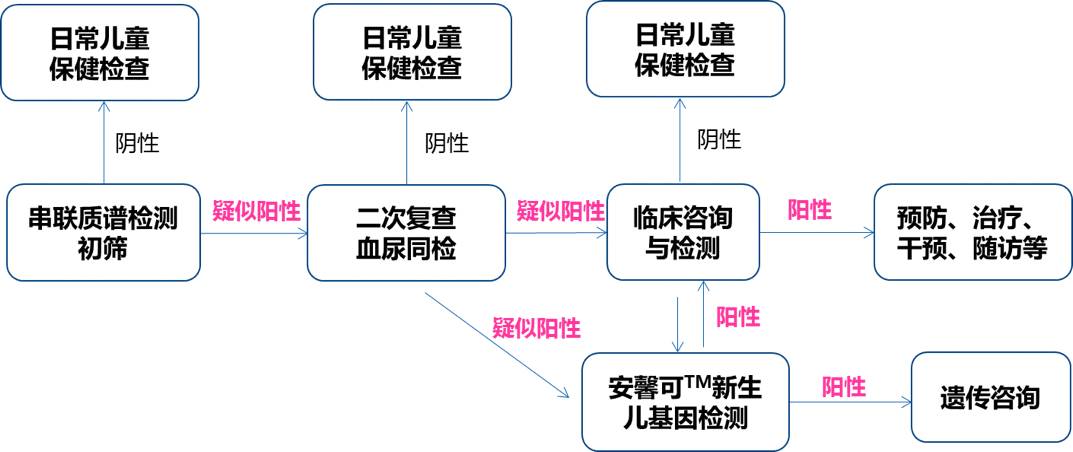
华大基因新筛“筛诊一体”流程示意
案例1
明明 (化名) ,8个月,通过华大基因新生儿遗传代谢病检测的“筛诊一体”流程,出生7天内采集了足跟血,新筛初筛结果异常。医务人员第一时间联系家属带孩子回原采样医院进行二次采血,华大基因提供了免费复查,但结果仍异常。医务人员再次电话通知家属带孩子去郑州大学第三附属医院进行确诊,同时华大基因免费为患儿提供了新生儿基因检测,最终临床和基因诊断结果一致为“甲基丙二酸血症”,之后家属谨遵医嘱、食用特殊配方奶粉、坚持治疗并定期随访复查,至今都未出现临床症状,且孩子的身体及智力发育均良好。
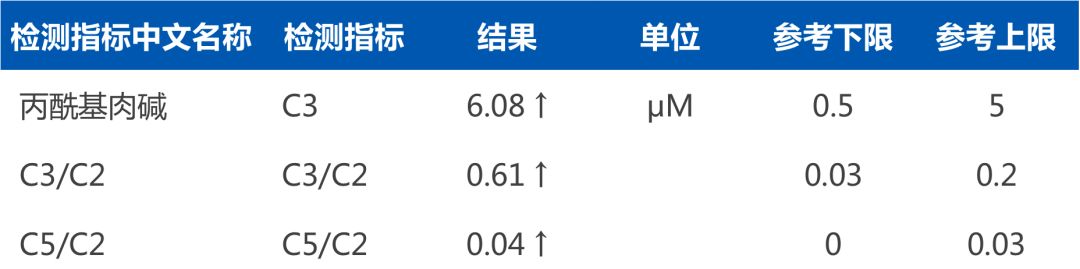

遗传代谢病串联质谱检测报告

新生儿基因检测-常见遗传病报告

健康成长的明明 (化名)
案例2
亮亮 (化名) ,2岁,通过华大基因新生儿遗传代谢病检测“筛诊一体”流程,确诊为“甲基丙二酸合并同型半胱氨酸血症”。第一时间的鉴别诊断为临床治疗提供了有效依据,不同于明明,亮亮不需要吃特殊配方奶粉,但也仍需坚持用药治疗。两年来家属遵医嘱、坚持治疗,并定期随访复查,至今未出现临床症状,且孩子的身体及智力发育均良好。
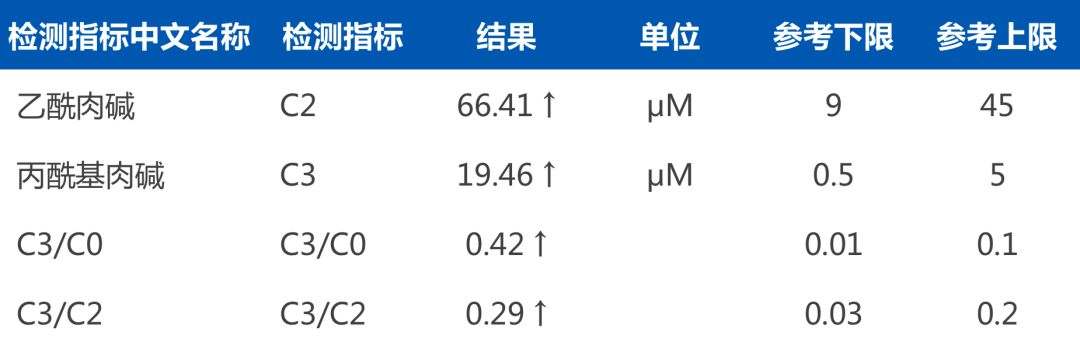

遗传代谢病串联质谱检测报告
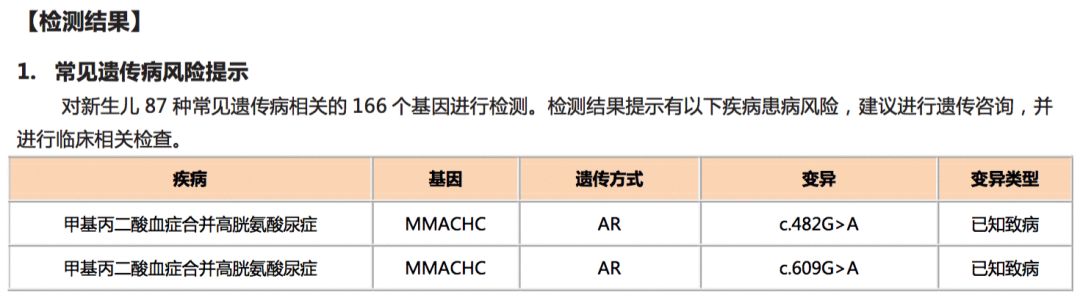
新生儿基因检测-常见遗传病报告
新生儿遗传代谢病“筛诊一体”闭环服务
2015年底,长垣县人民政府启动了免费基因检测健康筛查服务项目,并率先实现了新生儿遗传代谢病的“筛诊一体”闭环服务。
在长垣县政府、县卫计委的精心策划和细致组织下,将新生儿遗传代谢病“筛诊一体”的整体解决方案应用于惠民项目中,从知情同意、采样、送样,到报告发放、阳性通知,全流程设计周全清晰,执行顺畅到位,并且对已确诊的阳性患儿定期随访、及时关怀,在当地民众中取得了强烈反响。

长垣县政府免费基因检测健康筛查文件 (左)
新生儿遗传代谢病“筛诊一体”方案的流程宣传 (中、右)
对于新筛检测结果提示疑似阳性的新生儿,长垣县医务人员会第一时间电话通知其家长去复查或转诊。同时,华大基因客服人员也会定期电话回访家长,了解新生儿健康近况,尽可能确保每一位疑似阳性的新生儿都得到及时的复查或确诊,每一位确诊的阳性患儿都得到有效的治疗。

华大基因团队与医务人员跟进复查与确诊情况
截止到最新,在长垣已完成新筛检测的3万名新生儿中,累计确诊阳性12例。在确诊阳性病例中,除《新生儿疾病筛查技术规范》中明确要求必须开展的苯丙酮尿症 (PKU) 4例,甲基丙二酸血症远高于既往发病率数据,共发现3例。在基因检测诊断中发现有1例为意义未明的新发突变位点。惠民项目的落地执行不仅让当地百姓享受到基因科技的服务,体会到科技力量带来的温暖关怀,也推动了临床研究的进展。

—— Qian J, Wang X, et al. 2017 Aug 28
2017年,华大基因在《小儿内分泌与代谢》杂志上发表了《应用靶向基因高通量测序技术诊断基于串联质谱筛查的干血片疑似阳性样本的可行性研究》,详细描述了“筛诊一体”能够有效简化和缩短疑似阳性患儿的确诊流程。
有
效
干
预
传
递
温
暖
目前,华大基因新生儿遗传代谢病检测“筛诊一体”流程已经在安徽太和、青海海西州、青岛黄岛区、陕西商洛等多个城市和地区先行先试,且成效显著。我们希望,通过政府及各方社会力量的共同努力,将新筛的“筛诊一体”、新生儿相关检测的闭环服务在中国更大范围的积极推进,推动我国出生缺陷防治工作规范、有序开展,加强儿童重大疾病防治,提高我国的公共卫生经济效益,进一步构建和完善中国人的疾病遗传图谱,并为更多的普通家庭带来更及时的科学指导,愿祖国的下一代健康快乐成长。
△
关于甲基丙二酸血症
疾病概述
甲基丙二酸血症 (Methymalonic acidemia, MMA) 又称甲基丙二酸尿症,是先天性有机酸代谢异常中最常见的疾患,为常染色体隐性遗传病。据调查报告显示,中国台湾省患病率约1.2/10万;上海新华医院筛查76万例新生儿,患病率为3/10万;浙江省儿童医院筛查近13万例,患病率为1.5/10万。
——顾学范. 临床遗传代谢病. 北京:人民卫生出版社
发病机制
根据酶缺陷的类型,甲基丙二酸血症分为甲基丙二酰辅酶A变位酶 (Mut型) 自身缺陷或其辅酶钴胺素 (Cobalamin, cbl,维生素B12) 代谢障碍两大类。钴胺素代谢障碍包括6个类型,分别为cblA、cblB、cblC、cblD、cblF和cblH。Mut、cblA、cblB和cblH缺陷型仅表现为MMA,故称为单纯型MMA,cblC、cblD和cblF缺陷型则表现为MMA伴同型半胱氨酸血症 (Homocysteinemia, HCY),故称为MMA合并同型半胱氨酸血症。
两大类疾病均导致患者体内毒性代谢产物甲基丙二酸、3-羟基丙酸、甲基枸橼酸蓄积,线粒体能量代谢功能降低,引起以神经系统损害为主的多脏器损害。
临床表现
甲基丙二酸血症患儿多在出生后前几个月或几年的时间内发病,临床表现包括厌食、呕吐、张力减退、发育迟缓、厌恶高蛋白饮食、代偿失调等,少部分患者在早期就可发病,需在发生代偿失调的情况下及时治疗。甲基丙二酸血症的并发症包括智力障碍、小管间质性肾炎、基底神经节梗塞、胰腺炎、视神经萎缩等。
① 婴儿型:昏迷、呕吐、脱水等,发病期间肝肿大、张力减退、脑病等,还包括代谢酸中毒、阴离子间隙过高、酮症、酮尿、高氨血症、高甘氨酸血症、血小板及嗜中性粒细胞减少、新生儿败血症等;
② 轻微型/成人型:多数情况下尿甲基丙二酸水平偏高,表现正常,但易突发急性代偿失调。
(转化医学网360zhyx.com)
还没有人评论,赶快抢个沙发