 腾讯登录
腾讯登录PLOS ONE:甲型流感病毒进化和传播规律研究获进展
| 导读 |
甲型流感为急性呼吸道传染病,其病原体是一种新型的甲型H1N1流感病毒,在人群中传播。自2009年开始,甲型H1N1流感在全球范围内大规模流 行。2010年8月,世卫组织宣布甲型H1N1流感大流行期已经结束。2012年我国甲型H1N1流感病例超过300例,由于流感病毒经常发生抗原漂移, 因此经常发生规模大小不等的暴发流行。
中国科学院动物研究所何宏... |
甲型流感为急性呼吸道传染病,其病原体是一种新型的甲型H1N1流感病毒,在人群中传播。自2009年开始,甲型H1N1流感在全球范围内大规模流 行。2010年8月,世卫组织宣布甲型H1N1流感大流行期已经结束。2012年我国甲型H1N1流感病例超过300例,由于流感病毒经常发生抗原漂移, 因此经常发生规模大小不等的暴发流行。
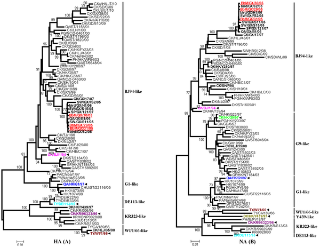
中国科学院动物研究所何宏轩研究员领导的野生动物疫病研究组2009年在《科学通报》上发表了甲型H1N1北美毒株特征(Chinese SciBull,2009),证明了甲型H1N1流感的变异和进化,对于人类呈现高亲和力、低致病性、不具有高致病性流感病毒的特性、对神经氨酸酶抑制剂类药物敏感,但对 金刚烷胺类药物具有抗性等4大特征。近期又对甲型流感爆发以来H1N1病毒所有片段进行了选择压力分析,结果发现除NS片段外其余7个片段的编码区都呈现 明显的负选择压力,表明它们对人是比较适应的。
NS片段是流感病毒的最小片段,其编码NS1和NS2两个蛋白,分析发现NS1片段也呈现负选择压力,而NS2片段则在流感刚刚爆发的4月呈现出明 显的正选择压力,因此认为NS片段极有可能在以前感染人H1N1病毒中从未出现过,在2009H1N1病毒中出现对于甲型流感进入人体并导致疫情爆发可能 具有一定的作用,而另一个不同寻常之处在于2009年8月的病毒NS2片段再次表现出明显的正选择压力,这与一般的病毒跨宿主传播的遗传进化理论不符。对 于这一奇怪现象,认为这与7月份以后甲流病毒传播速度和传播范围的急速扩大有关系,病毒接触到的群体数量、群体多样性都大为增加,因此病毒在进化过程中再 一次受到了正选择压力,进一步表明NS片段是2009年前后重组到人流感病毒基因组的可能性。
NS片段的遗传突变网络分析,甲型流感病毒自4月份出现以来,就以两个基础的突变型(G1型,123V 和G2型,123I)为原点进行进化,这个变异的位点处于NS1的编码区。出现这一现象有两种可能,一是在病毒最初传播到人宿主时就是以两个突变型的形式先后传入的,该突 变可能在甲流未传入人的时候就已经出现,因为这两个基本突变性出现的时间都非常早,同时又分别都有大量的感染群体和后期进化毒株的出现;二是病毒先以G1 或G2中的一种进入人体,而后另外一种由原始突变型进化而来,但显然该位点的突变使病毒获得了明显的感染传播优势,在短期内形成了庞大感染群体。
综上所述,认为NS片段是这次甲型流感暴发过程中的一个极为特殊的片段,很有可能对甲流爆发以及蔓延起到了重要的作用。这一研究结果发表在《PLOS综合》杂志上。该项目获得了国家自然科学基金青年基金,中科院知识创新工程等项目的支持。
原文链接:
Reassortant H9N2 Influenza Viruses Containing H5N1-Like PB1 Genes Isolated from Black-Billed Magpies in Southern China
H9N2 influenza A viruses have become endemic in different types of terrestrial poultry and wild birds in Asia, and are occasionally transmitted to humans and pigs. To evaluate the role of black-billed magpies (Pica pica) in the evolution of influenza A virus, we conducted two epidemic surveys on avian influenza viruses in wild black-billed magpies in Guangxi, China in 2005 and characterized three isolated black-billed magpie H9N2 viruses (BbM viruses). Phylogenetic analysis indicated that three BbM viruses were almost identical with 99.7 to 100% nucleotide homology in their whole genomes, and were reassortants containing BJ94-like (Ck/BJ/1/94) HA, NA, M, and NS genes, SH/F/98-like (Ck/SH/F/98) PB2, PA, and NP genes, and H5N1-like (Ck/YN/1252/03, clade 1) PB1 genes. Genetic analysis showed that BbM viruses were most likely the result of multiple reassortments between co-circulating H9N2-like and H5N1-like viruses, and were genetically different from other H9N2 viruses because of the existence of H5N1-like PB1 genes. Genotypical analysis revealed that BbM viruses evolved from diverse sources and belonged to a novel genotype (B46) discovered in our recent study. Molecular analysis suggested that BbM viruses were likely low pathogenic reassortants. However, results of our pathogenicity study demonstrated that BbM viruses replicated efficiently in chickens and a mammalian mouse model but were not lethal for infected chickens and mice. Antigenic analysis showed that BbM viruses were antigenic heterologous with the H9N2 vaccine strain. Our study is probably the first report to document and characterize H9N2 influenza viruses isolated from black-billed magpies in southern China. Our results suggest that black-billed magpies were susceptible to H9N2 influenza viruses, which raise concerns over possible transmissions of reassortant H9N2 viruses among poultry and wild birds.
来源:中科院动物所
还没有人评论,赶快抢个沙发